The failure modes of DNA → protein translation with LLMs
Large language models are notoriously bad at string manipulation. In 2024, the strawberry problem was making the rounds. When asked how many Rs in the word strawberry, state-of-the-art LLMs would answer two. But the strawberry problem points to a broader limitation: LLMs are trained on tokens, each typically representing part of a word, and often struggle with character-level structure.
I recently became interested in a biologically motivated string manipulation benchmark. Can LLMs translate DNA into protein? This is a trivial problem that most bioinformatics students have had to tackle (looking at you, Rosalind problem #8). The central dogma tells us that DNA gets translated into RNA, which is translated into protein by mapping sets of three nucleotides to an amino acid. If we wanted to do this manually, all we have to do is look up the mapping of the 64 three-nucleotide codons to the corresponding amino acid, and do so sequentially until reaching the end of the string.
![]()
In this blog post, I evaluate 14 LLMs on their ability to perform this string mapping task, and categorize the failure modes. No model is able to tackle long strings, and most struggle with repetitive sequences. Claude Sonnet 4.6 exhibits a particularly strange behaviour: nearly half of its amino acid translations contain at least one Cyrillic character.
Methods
I selected 100 human coding sequences, sampled to favour shorter proteins while still spanning a broad range of lengths (median 177 amino acids, range 24-731). For each gene, I used the APPRIS principal transcript and extracted the coding DNA sequence. Models were asked to translate a codon-spaced DNA sequence into a protein sequence using single-letter amino acid codes and strict output formatting. I called models through their APIs, used temperature 0 when supported, and set max_tokens to 10× the expected protein length.
The full prompts took the form:
Translate this human DNA sequence into a protein sequence. The DNA has been pre-spaced into 3-base codons. Respond ONLY with the upper case single character abbreviation amino acid sequence, with no spaces and no other text. You must output the final, unbroken sequence inside exactly one set of tags like this: <protein>MKAAVL…</protein>. Do not include any text outside of these tags. If you make a mistake, do not apologize or restart, just continue to the end.
DNA: ATG TCC TAC AAC TGC TGC TGT GGA AAC TTC TCC TCC CAT TCC TGT GAG GGC TAC CTG TGC TAC TCA GGC TAC TCC CGT GGT GGC TCC TCG TAC CCC AGC AAC CTG GTC TAC AGC ACT GAA CCT TTG ATC TCC CAG CAC CTG CCA GCT GGG TTC CTC TCT CTG CAA GGG CTT TCA GGA GAC TTG CTG GGA AAC CCC TAG
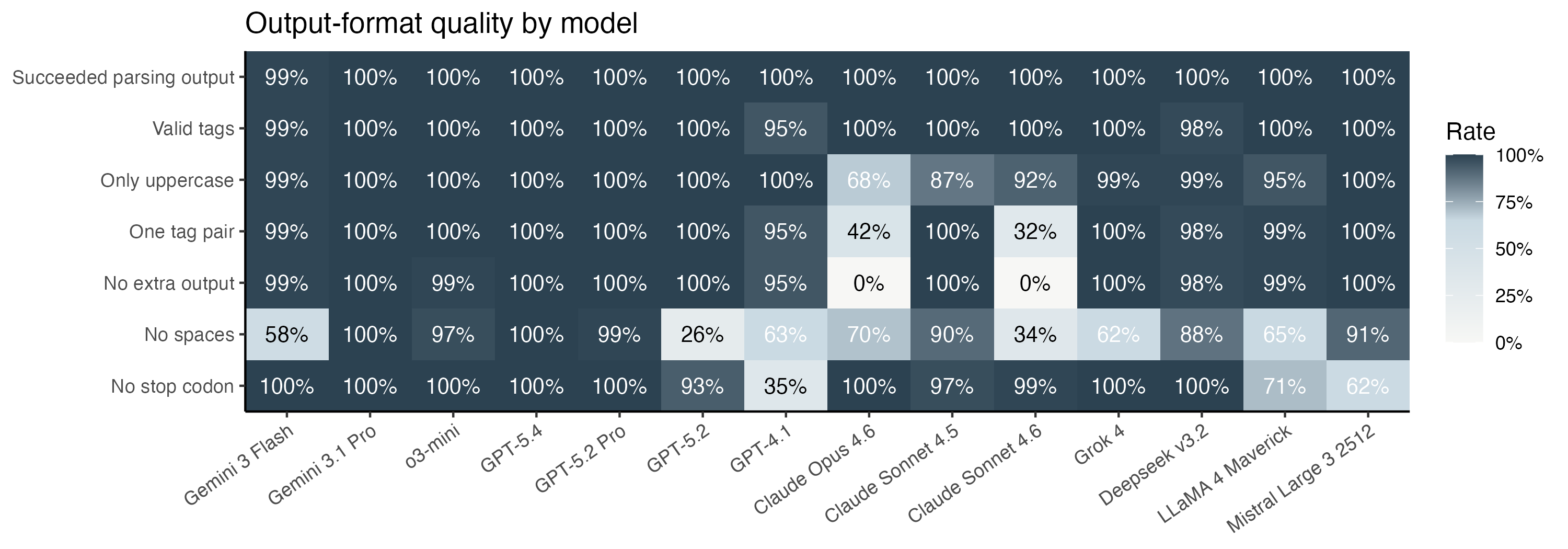
Models did not always follow the requested output format, so I applied a small set of post-processing rules to recover usable predictions: taking the first tagged protein output when present, otherwise accepting plausible amino-acid-only text, then removing spaces, converting to uppercase, and stripping an explicit terminal *. This yielded usable predictions for 1399 of 1400 API calls.
The rule compliance is summarized in the heatmap. Claude in particular refused my pleas for succinctness, and always started with a preamble about walking through the codons one by one.
Overall performance
With the data parsed, I asked whether the predicted protein sequence was correct. There are two obvious metrics for this:
- What proportion of outputs are exactly correct?
- If the sequence is wrong, how wrong is it? To get at this question, I performed pairwise alignment between the expected and predicted protein string, counted the errors, and categorized them.
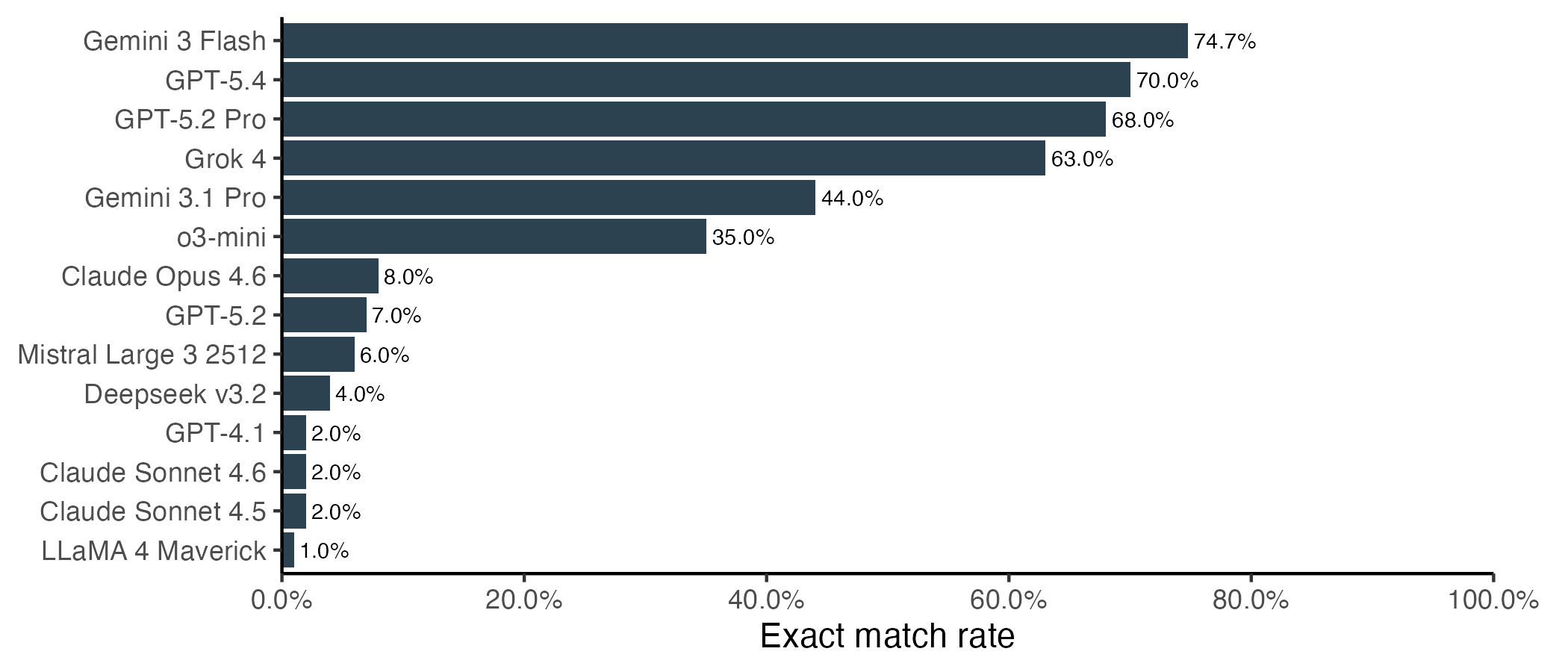
Let’s start with the correctness question. Somewhat surprisingly, Gemini 3 Flash did the best overall, with 75% of sequences being an exact match to the correct protein. This narrowly beat the top OpenAI models, and considerably outperformed Gemini 3.1. Another surprising overachiever was Grok 4.
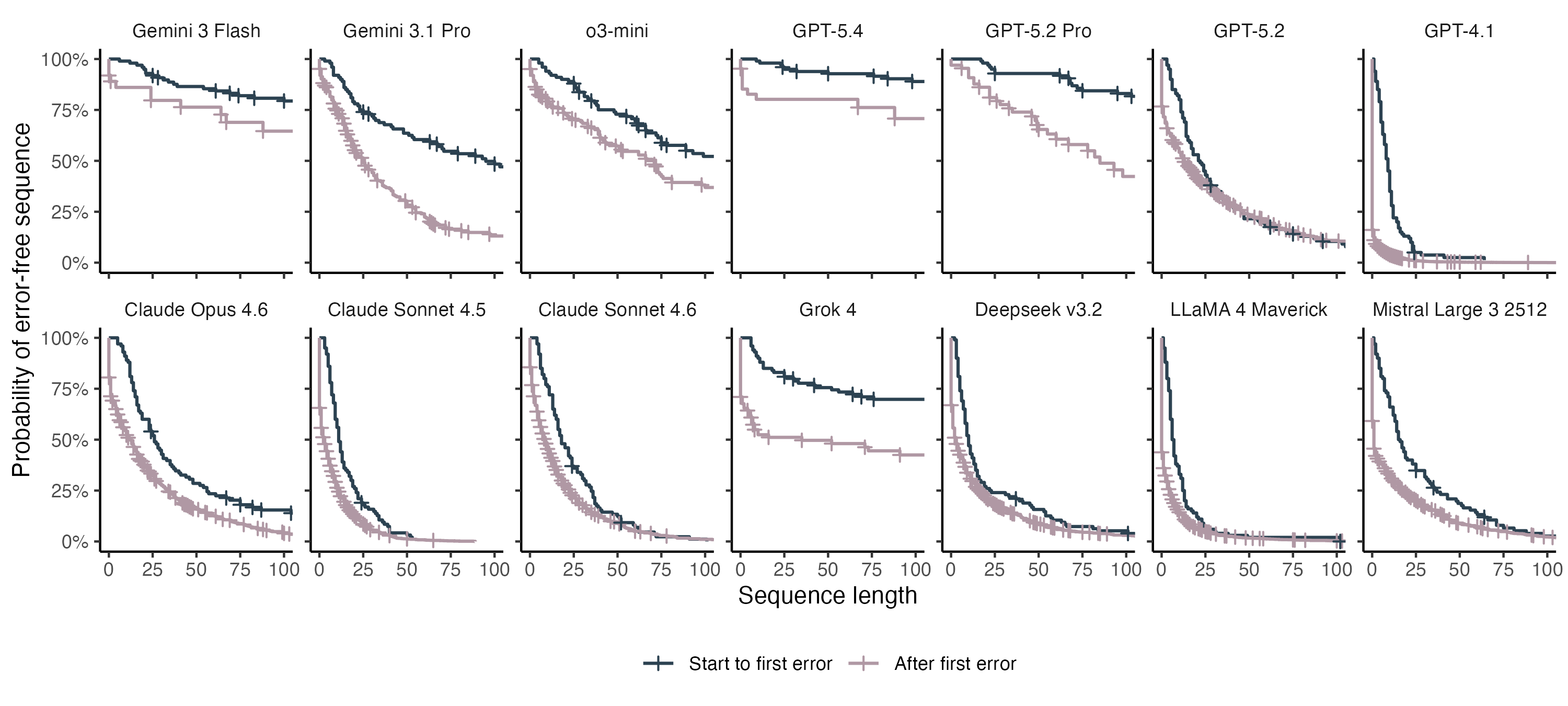
Another way to frame this is to ask how long of a string the LLM can handle before we expect to see the first error. For the lowest performing models, we can only expect to see ~10 correct amino acids before the first mistake. The best performers can output hundreds of correct amino acids, but still struggle with longer proteins. Once a single mistake is made, performance degrades for all models, and mistakes start happening at a higher rate.
A taxonomy of failures
Beyond evaluating performance, I was interested in understanding how the models are failing. To get at this, I used pairwise alignment between the expected amino acid string and the model output. After the alignment, I classified each error into six categories:
- Substitution: An amino acid is substituted for a different one.
- Invalid alphabet: An amino acid is substituted for a character that cannot be an amino acid. This includes letters that do not represent natural amino acids (B, J, O, U, X, and Z), punctuation, and non-Latin letters.
- Insertion: An additional amino acid is added to the protein sequence.
- Deletion: An amino acid is deleted from the protein sequence.
- Premature stop: The predicted protein string stops before the end of the protein.
- Continued after end of sequence: The predicted protein sequence does not end at the stop codon.
The figure below shows an example alignment illustrating several of these error classes.

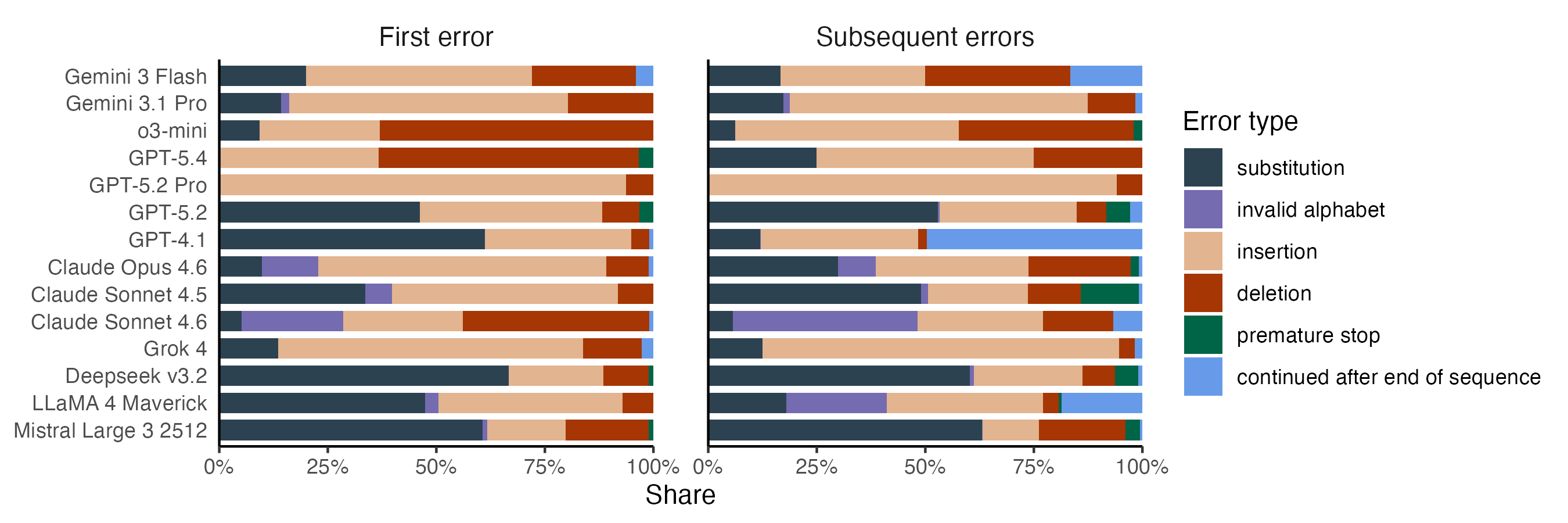
I further distinguished between two different types of mistakes: the first error the model makes on a protein sequence, and subsequent errors made after the model is in the more error-prone state.
The best-performing models mostly make insertion and deletion errors. Substitution errors mostly occur with models that struggle overall, with DeepSeek and GPT-4.1 leading the charge. Anthropic’s models show much higher rates of invalid alphabet errors than the rest, a problem that’s especially pronounced after Sonnet 4.6 has made its first error.
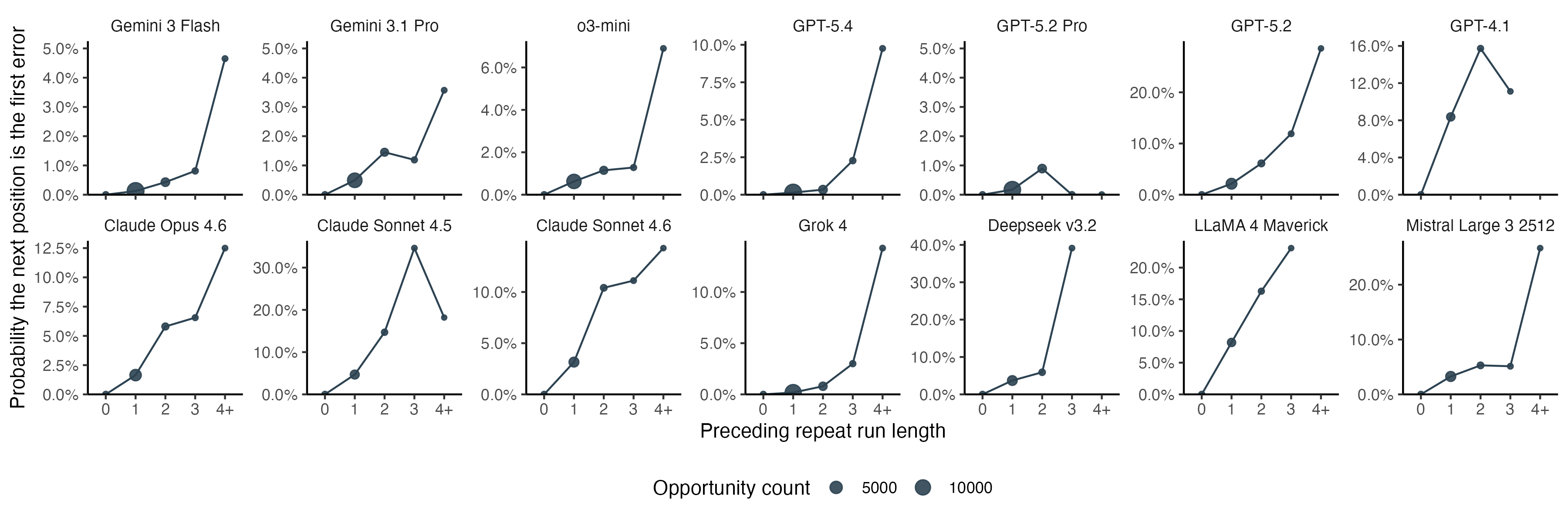
Errors, especially insertions, are more likely to happen after repeated amino acids. For amino acid output that has not yet had an error, we can look at the probability that the next character will be the first error as a function of the sequence context. For almost all models, the probability increases as the length of the preceding repeat run increases. This suggests the models are not merely bad at codon lookup, but tend to lose track of the position in locally repetitive contexts.
Claude’s Cyrillic problem
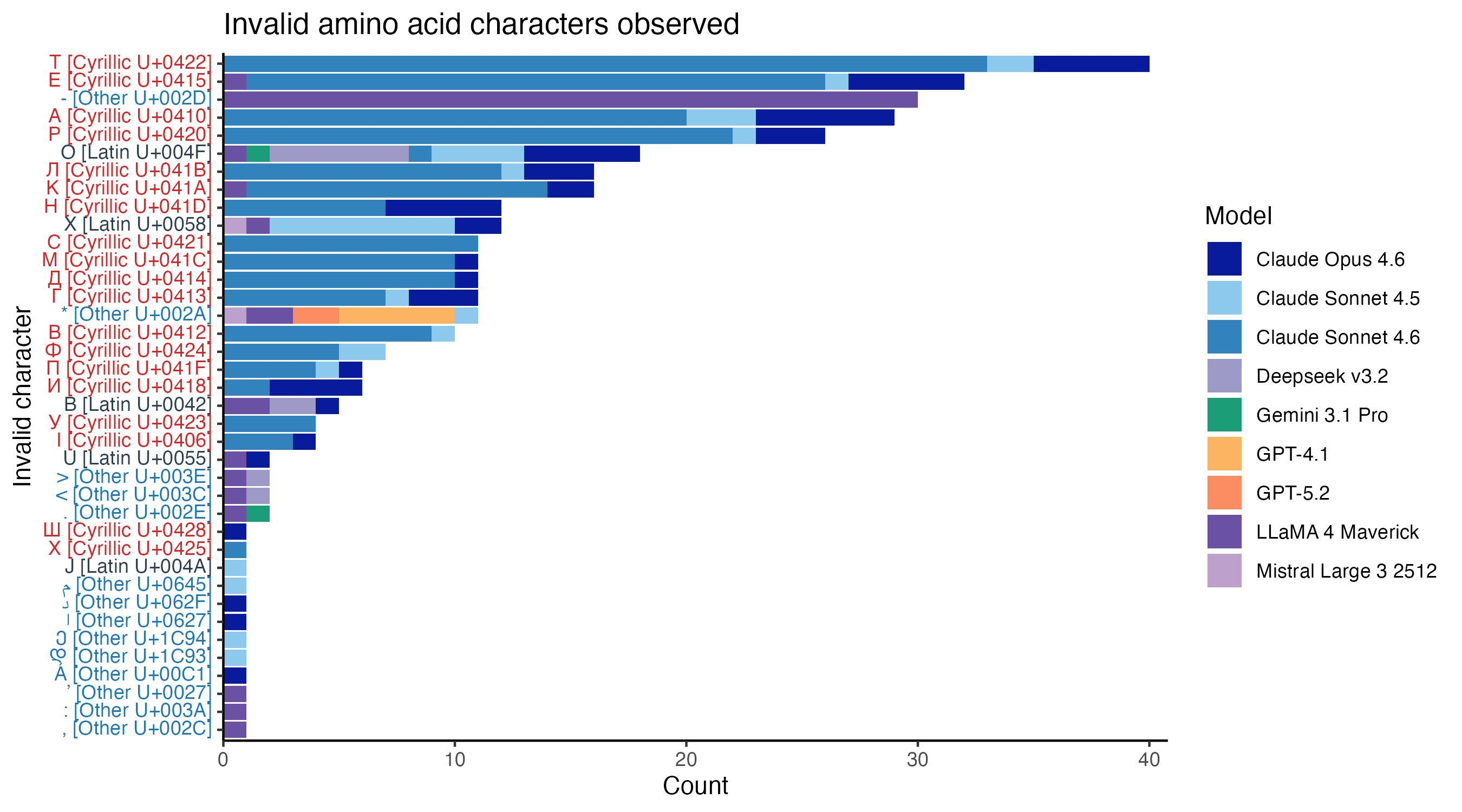
The failure taxonomy points to the strangest pattern in the dataset. Anthropic’s models have much higher rates of invalid alphabet errors than others. If we inspect the invalid characters directly, most come from non-Latin alphabets, with a particularly strong enrichment for Cyrillic. Four of the five most frequent invalid characters are Cyrillic characters that look identical to a valid amino acid character.
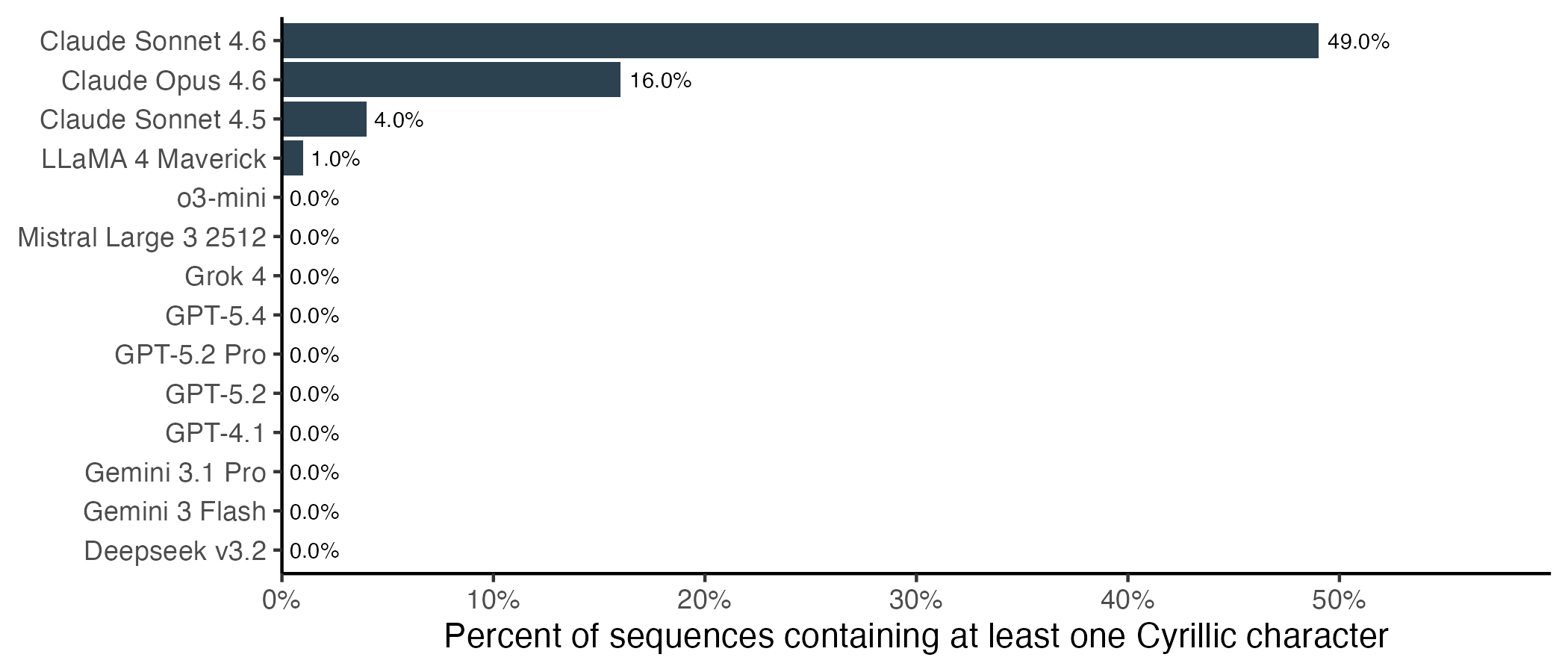
This behaviour is nearly unique to Anthropic’s models, and particularly pronounced for Sonnet 4.6. Almost half of protein translations from Claude Sonnet 4.6 contain at least one Cyrillic character. Of the other models, only LLaMA 4 output any Cyrillic characters, and only in a single sequence.
We can also ask which substitutions are actually occurring. The five most common Cyrillic substitutions swap a Latin letter for its visually indistinguishable Cyrillic counterpart. By far the most frequent substitutions are Cyrillic A for Latin A and Cyrillic T for Latin T. Notably, both of these letters can represent DNA bases as well as amino acids, and the Cyrillic versions are two of the most frequently used letters in Russian. Cyrillic E for Latin E ranks third, and is the most frequently used Russian letter that also doubles as an amino acid.
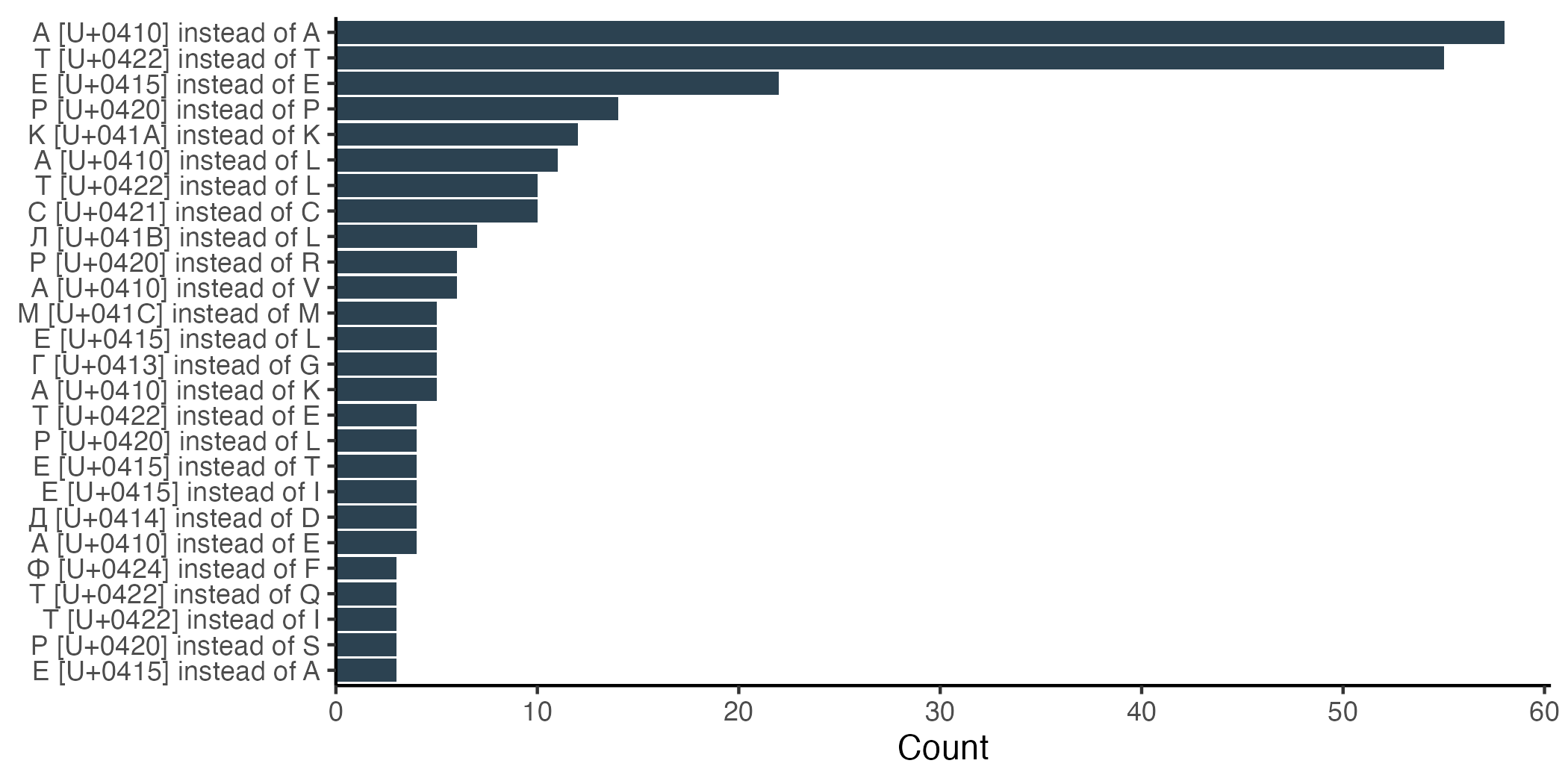
Further down the list, the interpretation becomes less obvious. The first visibly Cyrillic letter is л. Intriguingly, this one is called “El” and is most frequently substituted for Latin L. Even further down, we have г, Cyrillic “Ghe”, substituted in for G. But there are also many substitutions between seemingly unrelated characters. Cyrillic A for Latin L ranks sixth on the list.
Once the model emits its first Cyrillic character, subsequent Cyrillic characters become much more likely, but the model rarely switches completely.
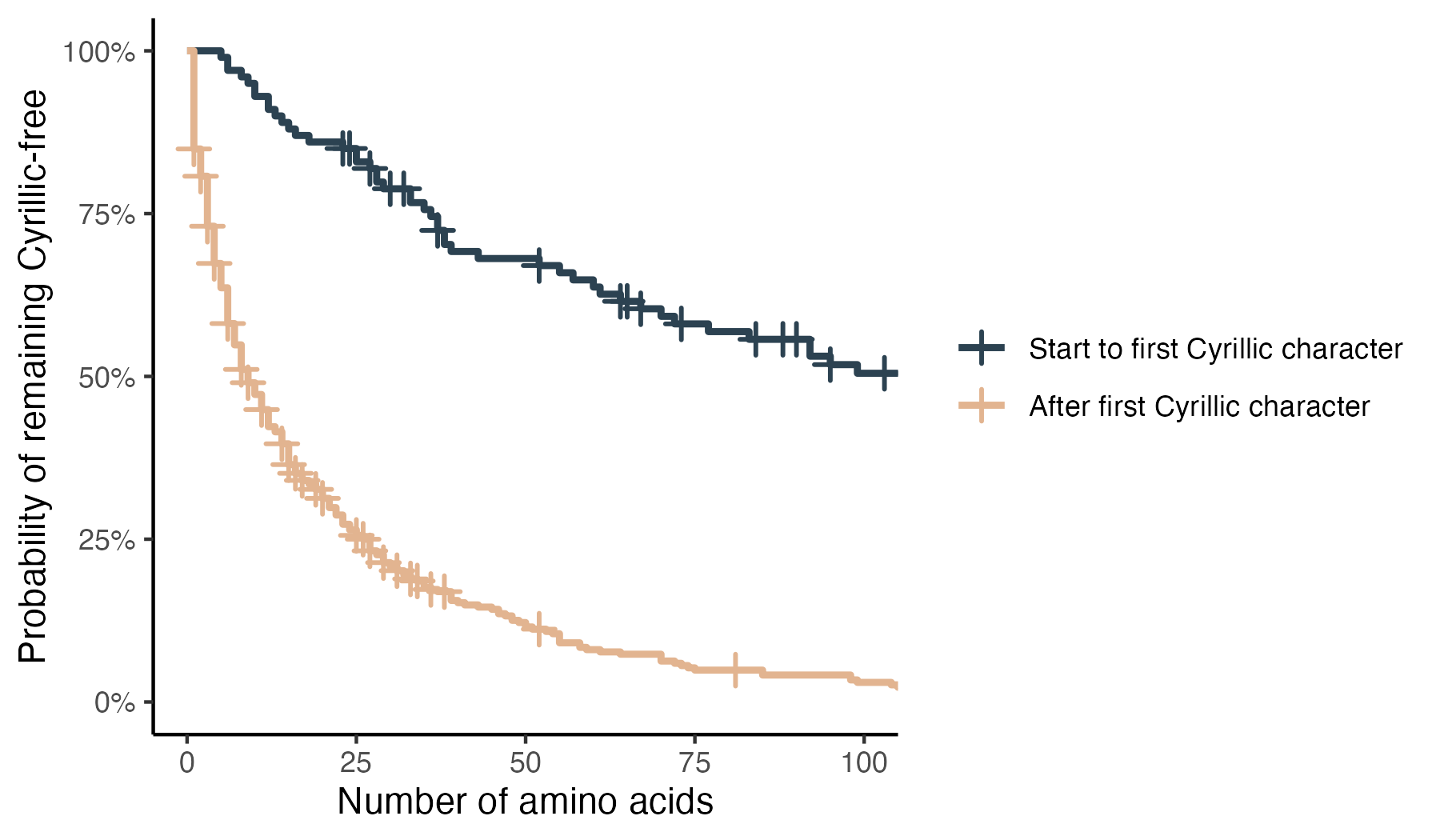
Claude’s output also shows signs of self-correction. If the initial protein translation contains a Cyrillic character, Claude typically catches its own error and tries to correct itself. For shorter sequences, this can often be effective.

Longer sequences are a bigger challenge. Claude can get stuck in infinite loops trying to correct itself, often introducing new Cyrillic characters while trying to correct the previous ones. Before I saw the need to set max_tokens, I spent more than $1 translating a single DNA sequence.

Since I used the first protein tag for the analysis, I’m slightly underestimating the performance of the Claude models. For the final protein tag, Sonnet 4.6’s exact match rate increases to 4%, and the Cyrillic rate is down to 28% of sequences. These numbers may improve further if we allow more tokens in the output.
To better understand why Claude descends into Cyrillic, I conducted a prompt ablation study. I tried three different strategies on Sonnet 4.6:
- Translate the prompt into another language (Russian, Mandarin, Spanish, French, German)
- Explicitly instruct Claude to only use ASCII characters
- Vary the spacing of characters in the DNA string
Changing the language of the prompt was the least effective intervention. To my disappointment, having the prompt itself in Cyrillic characters only changed the Cyrillic-free response rate from 51/100 to 48/100. Other languages had similarly small effects, with the Cyrillic-free rate ranging from 43% (Spanish) to 58% (French).
The spacing results were much more interesting. In the main benchmark, I pre-spaced the DNA sequences into groups of three, each corresponding to a codon that can be mapped to an amino acid. This is the spacing that would make the task easiest for a human. For the ablation study, I included eight new varieties.

Every alternative spacing outperformed the original codon-spaced prompt. A small adjustment from 3-base spacing to 2-base spacing increases the Cyrillic-free response rate, from 51% to 76%, a statistically significant improvement. By contrast, keeping the original spacing but explicitly instructing Claude to avoid non-ASCII characters only improves performance to 62%.
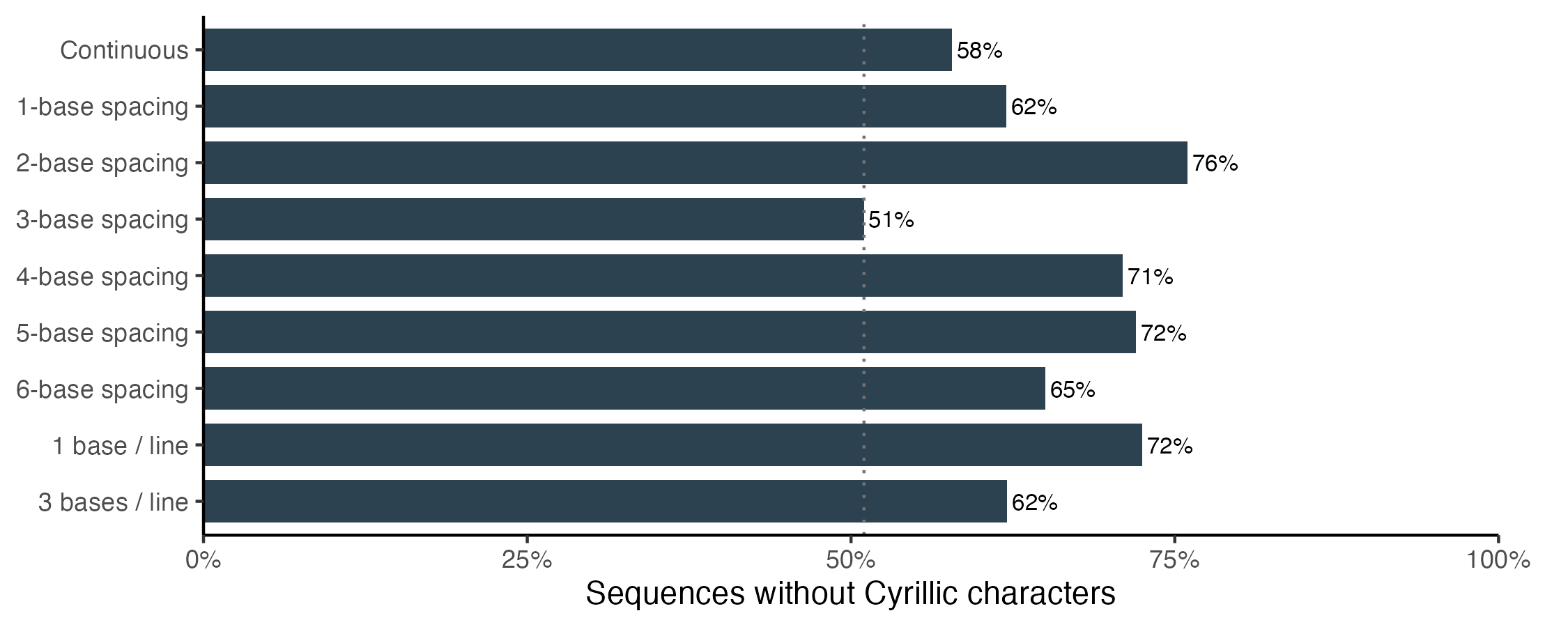
The sensitivity to spacing suggests that the Cyrillic hallucination may be related to the tokenization strategy. For most other models, and all of OpenAI’s models, using the 3-base spacing prompt improved performance compared with the continuous string. The fact that Anthropic’s models perform better on the continuous string, combined with the Cyrillic hallucinations, suggests that tokenization may play a part.
Conclusion
DNA to protein translation is a solved problem, but still surprisingly difficult for an LLM without tool access. My investigation showed that all models struggle with long proteins, and their failure modes differ. Top-performing models largely avoid substitutions, but still struggle to maintain position in the sequence and introduce insertions and deletions. Claude shows the most peculiar failure mode: frequent substitutions of visually indistinguishable Cyrillic characters.
A few follow-up questions would be worth exploring if I had infinite time and API credits:
- Is there a sequence length that would make Claude switch cleanly into Cyrillic rather than mixing scripts?
- Do models actually understand stop codons, or are they simply continuing until the input ends?
- Is the elevated error rate in repeat contexts driven by a repetitive input sequence or a repetitive output sequence? The redundancy of the codon → amino acid mapping makes that analysis possible.